We are introducing a completely new approach for treatment of inflammatory diseases based on the cross-sectional research areas of DNA repair and inflammation. We have demonstrated proof of principle showing that therapeutic treatment with our tool compound, an OGG1 inhibitor, TH5487 significantly reduces the inflammation in mice with lung inflammation.
Upon exposure to inflammatory agents, receptor-ligand interactions in cells cause an elevation of reactive oxygen species (ROS) and DNA damage. The pro-inflammatory immune cells that drive auto-immunity and inflammation suffer from high level of oxidative stress and therefore require specific detoxification enzymes for survival and function. One enzyme of specific interest is OGG1, 8-oxo guanine DNA glycosylase 1. Through binding to promoter regions, enriched in oxidized guanines, this enzyme recruits other proteins such as transcription factors, e.g. NF-ƘB to form complexes, promoting transcription of pro-inflammatory genes. Our OGG1 inhibitor TH5487 prevents OGG1 from binding to DNA and thereby dampening the inflammatory responses in animal models.
For more information about the mechanism of action please refer to the original publication.
We are advancing our leading science on the OGG1 pathway to develop novel therapies for severe lung inflammation diseases with large medical need such as ARDS, COPD, severe non allergic asthma, and idiopathic pulmonary fibrosis (IPF).
The Helleday lab is also open for collaborations and partnership to enable development of OGG1 inhibitors as potential anti-inflammatory therapies. Please contact Thomas Helleday directly for further information.
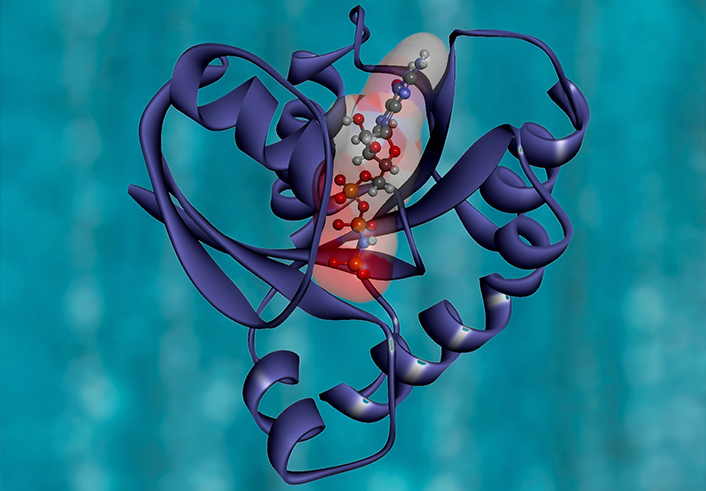
Left: Gallery illustrating the binding mode of 8-oxoguanine modified double stranded DNA to OGG1 (PDB: 1EBM); red to blue color coding of the OGG1 protein indicates a strong positive electrostatic potential of the binding site (blue = positive, red = negative); Later the gallery shows the crystal structure of OGG1 with TH5675, confirming targeting of the positively charged active site; Right: A video generated to demonstrate inhibitor binding and replacement of DNA as a ligand.
Lead generation of OGG1 inhibitors beyond TH5487
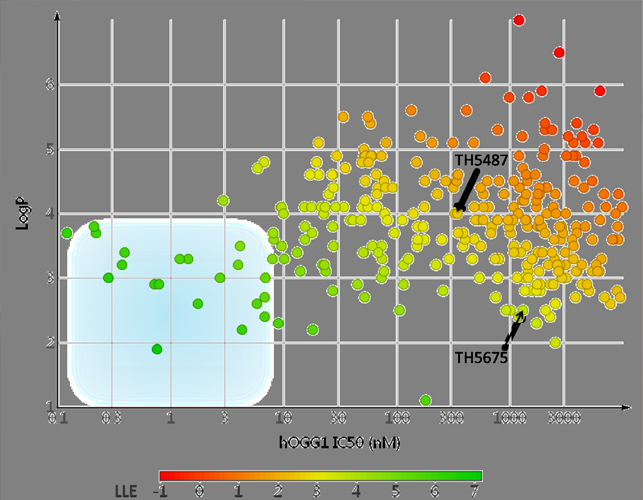
Initially, CBK149850 (45% inhibition at 10 µM) was identified as a starting point for lead generation from a high-throughput screen of 17,940 compounds. Interrogation of the structure-activity relationships (SARs) by systematic modification of CBK149850 yielded TH5487 as an initial lead molecule. While substantially more potent (IC50 = 342 nM), obtaining co-crystal structures proved very challenging, due to the limited solubility of both the inhibitors and the protein construct used. In the absence of adequate structural information the SAR of TH5487 was further explored, and collectively over 700 structural analogues were synthesized and tested biochemically. A real breakthrough came when a crystal structure was obtained of mouse OGG1 in complex with TH5675. The mouse homologue of OGG1 has a nearly identical active site but is more soluble due to solubilizing mutations at the protein surface. Furthermore, changing from TH5487 to the less potent but substantially more soluble TH5675 yielded crystals clearly showing bound ligand in the protein active site. To our surprise TH5675 bound with the hydrophobic p-iodophenyl moiety inserted deeply into the 8-oxoguanine pocket, while the benzimidazolone moiety was oriented outward and partially solvent-exposed. With this detailed atomic map at hand, tailored modifications could be made to further improve solubility and metabolic stability while simultaneously improving potency to low nanomolar digits and below. This is illustrated in the figures of the gallery on the left side, where a representative set of OGG1 inhibitors from the TH5487 series are showing progression from TH5487 towards optimal lipophilic ligand efficiency (LLE*) as indicated by the blue region. The second figure further demonstrates the benefits of structure based drug design, as compounds of the TH5487 series synthesized after access to the high resolution crystal structure are highlighted in green and those from before in red.
* for a review see: Leeson PD, Empfield JR (2010) Reducing the Risk of Drug Attrition Associated with Physicochemical Properties. Annu Rev Med Chem 45, 393-407.
This work was funded by the National Institute of Allergic and Infectious Diseases NIAID/AI062885 (I.B.), The Faculty of Medicine at the Norwegian University of Science and Technology and the Central Norway Regional Health Authority (A.S. and H.E.K., project no. 46056921), Svanhild and Arne Must’s Fund for Medical Research (A.S. and H.E.K.), Vinnova (A.C.-K and T.H.), the European Union’s Horizon 2020 research and innovation program under the Marie Sklodowska-Curie grant agreement no. 722729 (B.M.F.H. and T.H.), the European Research Council (T.H. TAROX Programme), The Knut and Alice Wallenberg Foundation and the Swedish Foundation for Strategic Research (T.H. and P.S.), Swedish Research Council (T.H. and P.S.), Swedish Cancer Society (T.H. and P.S.), the Swedish Children’s Cancer Foundation (T.H.), the Swedish Pain Relief Foundation (T.H.), and the Torsten and Ragnar Söderberg Foundation (T.H.). This project received funding from the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation programme grant agreement No 957495 DOIFF.
While these approaches cannot be overrated in their applications, the technologies have not yet reached their full potential in the field of medicine. On the one hand, organocatalysis in living cells would either be protein independent or taking place in the active site of enzymes, with the latter requiring small molecule activation to exist as an orthosteric technology. On the other hand, enzymatic mutations to improve functions within cells would require either short-term translation from for example an RNA-vaccine or permanent and legally challenging CRISPR/Cas methods.
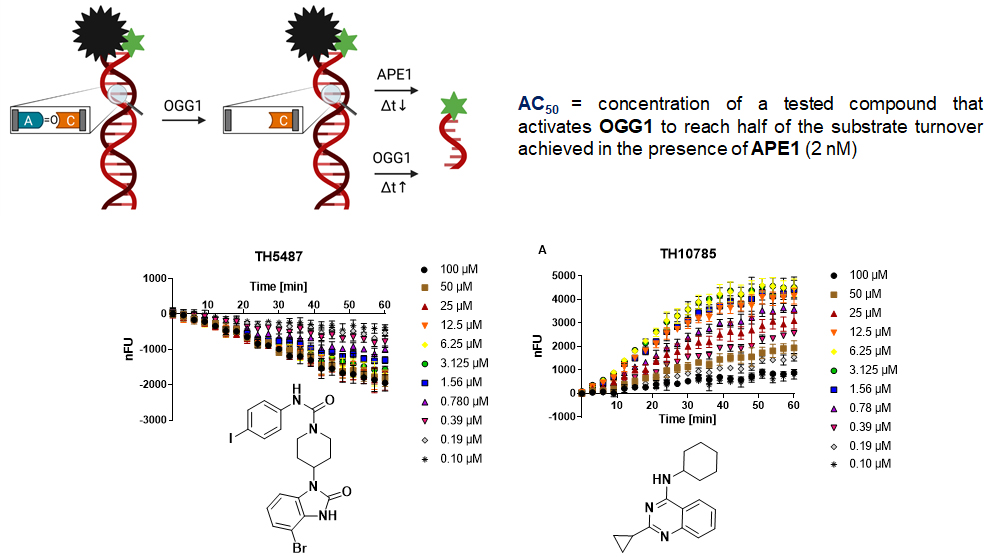
AC50 vs. IC50: TH10785 activates OGG1 function dose-dependently. The assay principle of the OGG1 biochemical assay releases a fluorophore after enzymatic excision of an 8-oxo-purine base. This can be coupled with APE1 for increased turnover or left incubating for slow turnover. Due to OGG1’s rudimentary AP-lyase function the latter version will still lead to full conversion after, just after a longer time frame. These two versions, with and without APE1, are suitable to either identify OGG1 inhibitors (w/ APE1, TH5487) or OGG1 activators (w/o APE1, TH10785). TH10785 has an AC50 of around 700 nM.

Reaction products of mono- and β,δ-bifunctional DNA Glycosylases have distinct repair pathways. Due to its weak AP-lyase function, OGG1 acts as a monofunctional DNA glycosylase in cells. Thus, reaction products are dependent on APE1. Other members of the DNA glycosylase family of enzymes possess β,δ-eliminating properties which require the action of PNKP1 instead. The OGG1 activator TH10785 removes protons from the DNA sugar-backbone, inducing an artificial PNKP1 dependency. At the same time, this reaction is so efficient, that 8-oxo Guanine lesions are neglected and abasic sites are preferred. In cells, the amount of abasic sites dwarfs those of 8-oxo Guanine, allowing TH10785 to be an efficient DNA Glycosylase-AP Lyase switch. In addition, and in contrast to inhibitors where large concentrations are required, low concentrations of activator may be efficient to double or triple the enzymatic function.
We now have published an initial answer to these challenges in a recent manuscript in the Journal Science. We describe the mode of action of the first chemist-synthesized organocatalyst that acts within a proteins active site. The DNA repair enzyme OGG1 or 8-oxo Guanine DNA Glycosylase 1 is boosted with regard to an originally rudimentary function. The increased AP-lyase function reprograms substrate preference from 8-oxo Guanine to abasic sites and further creates a new reaction product. This allows for an increase in oxidative DNA damage repair initiation. At the same, however, this accumulates downstream repair intermediates which are potentially lethal to cells with defective repair systems. When using different classes of the organocatalysts a rational overload or rescue of DNA damage may be achieved. In cells this has shown to work with PNKP1 inhibition. Here, DNA damage directed along the axis 8-oxo Guanine/abasic sites/single strand breaks/double strand breaks sensitizes for defects in DNA damage response. Read our full story here or reach out for more information and collaborate with us. We will continue to build on this discovery and develop workflows for broad development of enzyme targeted organocatalysts – which we have termed Amplizymes.
Situated centrally between all teams, the platform connects the streams of this new technology and establishes the necessary protocols. We are active within the data-driven life science where our platform of choice KNIME enables us to identify future enzyme candidates or target families. Based on a number of descriptors such as enzymatic reactions, substrate scope and pocket size we then rationalize the production of mutants and wild-type enzyme with the biochemistry team. Computational methods are also used to assess the suitability of a compound to perform organocatalysis. Basic determination of pKa, protein-ligand-interactions and functional group tolerance using docking are complemented with quantum-mechanical approaches to identify reaction pathways and perform molecular dynamics calculations. The latter is conducted in collaboration with the Himo lab at Stockholm University. A cornerstone, (Bio)conjugate chemistry uses (glyco)peptides, fluorophores, cleavable linkers and targeting moieties to control delivery, release and potency in cell-compartments which we investigate with the basic science team. Synthesis of Amplizymes expands classical medicinal chemistry, demanding a catalyzing atom center. Here, the necessary method development covers protection group, heterocyclic, photocatalytic and flow chemistry.
Together with biochemistry and in-vitro pharmacology teams, we assess target engagement, potency, substrate scope and kinetic parameter within enzymatic activity assays. Further, to enable substrate and product identification we drive the development of fluorescent, HPLC and gel-based assays for our in-house platforms on DNA glycosylases and the NUDIXes. The in-vivo pharmacology team and UDOPP inform for ADMET properties during the optimization of suitable compounds. We collaborate with the Stenmark lab (Stockholm University) for X-ray structures, the Hertweck lab (Leibniz Institute Jena) for mass spectrometry and the de Vega lab (CBM Severo Ochoa Madrid) for in vitro reconstitution of DNA repair pathways. Translationally we are engaged with the Stolz lab (Goethe University Frankfurt) for autophagy and cellular health screens, the Perona lab for Telomere biology (Alberto Sols, Madrid) and the Research Institutes of Sweden in Södertälje and SINTEF in Trondheim for industrial use of our technology.